Who is considered a “Sponsor” or a “Sponsor-Investigator”?
Sponsor: An individual, company, institution, or organization that takes responsibility for and initiates a clinical study involving an investigational product, but does not conduct an investigation.
Sponsor-Investigator: An individual who both initiates and actually conducts, alone or with others, a clinical investigation, and under whose immediate direction the investigational device or drug is administered, dispensed, or used. The term does not, for example, include a corporation or agency. The obligations of a sponsor-investigator include those of an investigator and those of a sponsor.
For additional definitions, refer to the University Sponsor and Sponsor-Investigator IND/IDE and FDA Pre-Submission Requirements policy.
University of Minnesota Sponsor and Sponsor-Investigator Policy
The University of Minnesota has accountability obligations for all University-sponsored and sponsor-investigator drug, device, or biological research and manufacturing at the University. The primary objective is to promote compliance with Food and Drug Administration (FDA) Investigational New Drug Application (IND) and Investigational Device Exemption (IDE) regulations (both Significant and Non-Significant Risk) that apply when individuals serve in the role of sponsor and/or sponsor-investigator. This policy does not apply to research sponsored by an external entity such as a commercial drug or device company, nor to Emergency Use INDs/IDEs.
When individuals serve as a sponsor or sponsor-investigator, they assume the same sponsor obligations under FDA regulations as a commercial drug or device company. Individuals who serve as sponsors or sponsor-investigators must also comply with applicable UMN requirements.
Link to the full policy: Administrative Policy: University Sponsor and Sponsor-Investigator IND/IDE and FDA Pre-Submission Requirements
Document Policy Compliance Through Completion of Form HRP-1899
HRP-1899 ensures that investigators understand and attest to all of the responsibilities of serving as a sponsor or sponsor-investigator. This form should be used throughout the lifecycle of the project to ensure that both initial and ongoing responsibilities are met.
Register Your Proposed Project with the Human Research Protection Program
The proposed project must be registered with the Human Research Protection Program (HRPP) during the new study submission in ETHOS. Registration of your IND or IDE/NSR IDE requires submitting your research to the IRB, and uploading form HRP-1899 in the “Attach files” section of the ETHOS Drug or Device page. See screenshot below.
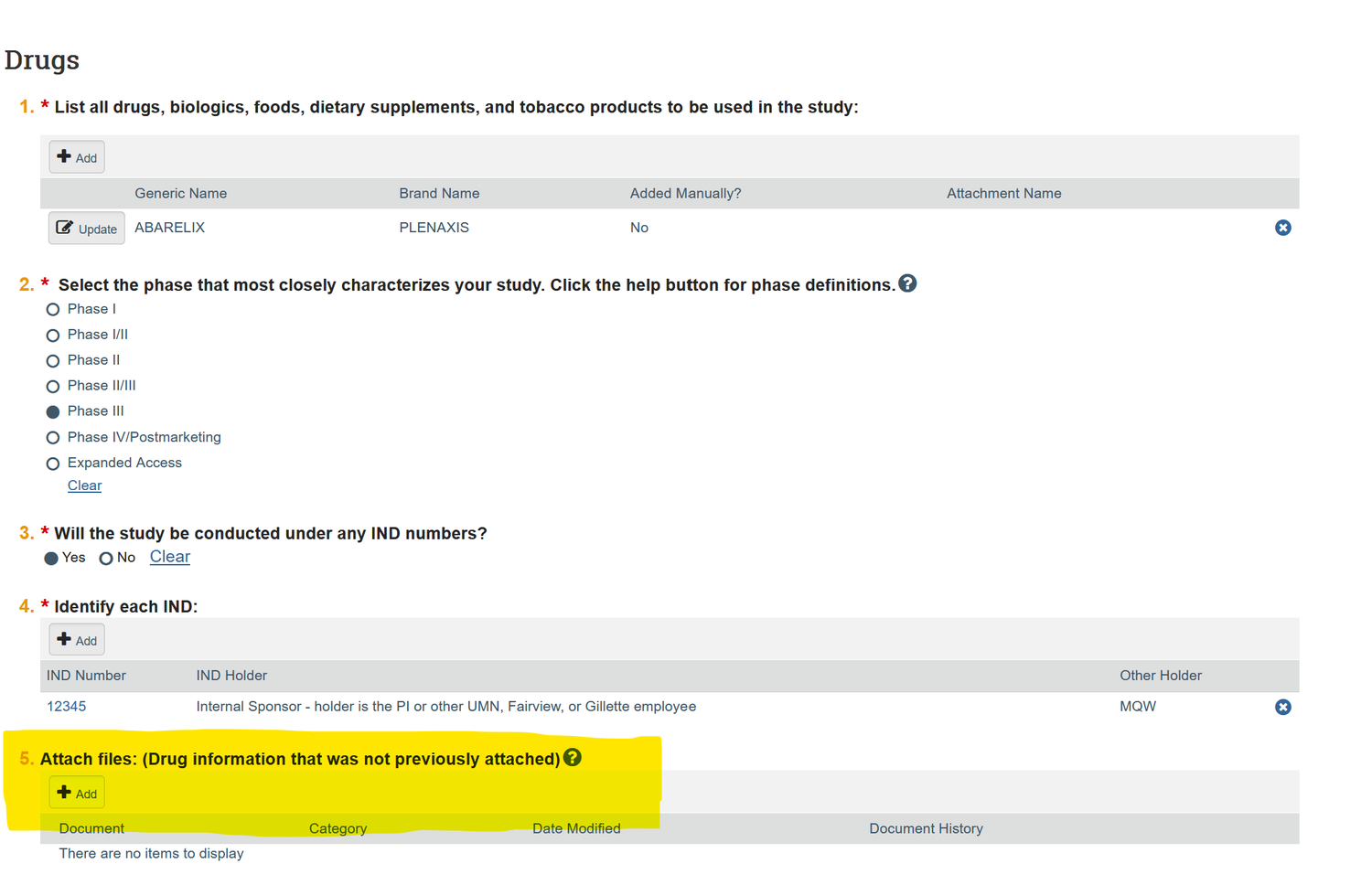
IND/IDE Central File Requirements
For each protocol, whether Non-Significant Risk IDE, Significant Risk IDE, IND, or an FDA pre-submission, sponsors and sponsor-investigators must submit the following to the University’s IND/IDE Central File Submission portal, as applicable:
- IND/IDE exemption or determination requests
- IND/IDE pre-submission and application
- Safety reports
- Amendments
- Communications including transcriptions of calls with the FDA
- Annual Reports
Central File Submission Portal
Click the link below to access the Central File submission portal. Use this link to submit your initial application to the FDA as well as all subsequent communication.
Central File Submission Portal
Monitoring Requirements
These projects require a robust monitoring plan. All IND, IDE, and NSR IDE studies with a University sponsor or sponsor-investigator must either utilize clinical trial monitoring services provided by the University through the Clinical and Translational Science Institute (CTSI); or provide a monitoring plan that is confirmed equivalent by CTSI. Please note that the FDA requires that sponsors select a monitor qualified by training and experience to monitor the progress of the investigation. Monitoring information should be clearly described in the IRB protocol. Eligibility for CTSI monitoring will be documented in the CTSI monitoring ancillary review; it is the responsibility of the study team to reach out to establish support.
Training Requirements
Training requirements for sponsors and sponsor-investigators mirror the requirements for principal investigators. These include:
- “Good Clinical Practice and Human Research Protections for Biomedical Study Teams–Basic Course”. This must remain up-to-date, and refresher courses must be completed every three years.
- HIPAA Privacy Training (2024)
- Responsible Conduct of Research (RCR) Core Training
For more information regarding training requirements and access to training modules, please refer to the IRB Education & Training webpage.
Ancillary Review
All research involving sponsor or sponsor-investigator initiated INDs/IDEs/NSR IDEs will undergo the following two ancillary reviews (among others) during the IRB review process.
Drug/Device Guidance/Regulatory Review: This review is prompted for all studies involving evaluation of drugs, devices, biologics, tobacco or dietary supplements.
Sponsor/Investigator Policy Compliance: This review is also prompted for all studies involving evaluation of drugs, devices, biologics, tobacco or dietary supplements. The purpose of this additional review is to ensure compliance with the policy, by checking that the aforementioned requirements are met. This review is also designed to ensure accuracy of the information provided in ETHOS.